
- +86-13363869198
- weimiaohb@126.com

Apr . 01, 2024 17:55 Back to list
drugs business API Manufacturing Performance Analysis
Introduction
Pharmaceutical Active Pharmaceutical Ingredients (APIs) represent the core chemical entities responsible for the therapeutic effect of drug products. Within the broader pharmaceutical supply chain, API manufacturing occupies a critical position, demanding stringent quality control, process validation, and adherence to complex regulatory frameworks. This guide details the scientific principles, manufacturing processes, performance criteria, potential failure modes, and industry standards governing API production, focusing on solid oral dosage form ingredients. The industry currently faces challenges regarding supply chain security, cost pressures, and the increasing complexity of novel therapeutic molecules. API quality directly impacts drug product efficacy, safety, and patient outcomes, making a thorough understanding of these parameters paramount for pharmaceutical manufacturers, procurement specialists, and regulatory bodies.
Material Science & Manufacturing
API manufacturing commences with raw material selection, typically sourced from petrochemicals, fermentation processes, or natural extraction. Common raw materials include benzene, toluene, and various carbohydrates. The synthesis pathway involves a series of chemical reactions, often multi-step, to transform these precursors into the desired API molecule. Critical process parameters (CPPs) like temperature, pressure, pH, and reagent stoichiometry must be meticulously controlled. Manufacturing methods vary depending on the API’s properties. Common processes include chemical synthesis (batch and continuous flow reactors), fermentation (for biologics and some small molecules), and purification (crystallization, distillation, chromatography). Crystallization is crucial for achieving desired particle size distribution (PSD) and polymorphism, impacting bioavailability. Polymorphism refers to the ability of a solid material to exist in more than one crystalline form. Each form exhibits unique physical properties such as solubility and melting point. Control of polymorphism is vital to ensure consistent drug product performance. The materials’ chemical compatibility with reactor materials (e.g., stainless steel, glass-lined steel) is essential to prevent corrosion or contamination. Scale-up from laboratory synthesis to commercial production requires careful consideration of heat transfer, mixing efficiency, and mass transfer limitations. Impurity profiling and control are also integral to the manufacturing process, demanding advanced analytical techniques.
Performance & Engineering
API performance is fundamentally tied to its physicochemical properties. Solubility, dissolution rate, permeability, and stability are key determinants of bioavailability – the fraction of administered drug that reaches systemic circulation. Particle size significantly affects dissolution rate, with smaller particles generally exhibiting faster dissolution. Hygroscopicity (tendency to absorb moisture) can impact API stability and flowability during manufacturing. Mechanical properties, such as bulk density and tapped density, are crucial for ensuring consistent tablet compression and capsule filling. Environmental resistance testing assesses API stability under various conditions (temperature, humidity, light exposure) to determine shelf life. Force analysis is used to evaluate the robustness of manufacturing processes, such as milling or granulation, to prevent particle breakage or deformation. Compliance with pharmacopoeial standards (USP, EP, JP) is mandatory, dictating acceptable limits for impurities, residual solvents, and other quality attributes. Analytical methods employed for performance evaluation include high-performance liquid chromatography (HPLC), gas chromatography (GC), differential scanning calorimetry (DSC), and X-ray powder diffraction (XRPD). Engineering controls, such as cleanroom environments and validated equipment, are essential to minimize contamination and ensure product quality.
Technical Specifications
| API Name | Molecular Weight (g/mol) | Purity (%) | Particle Size (µm) – D90 |
|---|---|---|---|
| Ibuprofen | 206.29 | >99.5 | 10-20 |
| Paracetamol (Acetaminophen) | 151.17 | >99.8 | 5-15 |
| Aspirin (Acetylsalicylic Acid) | 180.16 | >99.7 | 20-40 |
| Atorvastatin Calcium | 585.38 | >99.0 | 50-100 |
| Metformin Hydrochloride | 165.63 | >99.5 | 10-30 |
| Simvastatin | 418.58 | >99.2 | 20-60 |
Failure Mode & Maintenance
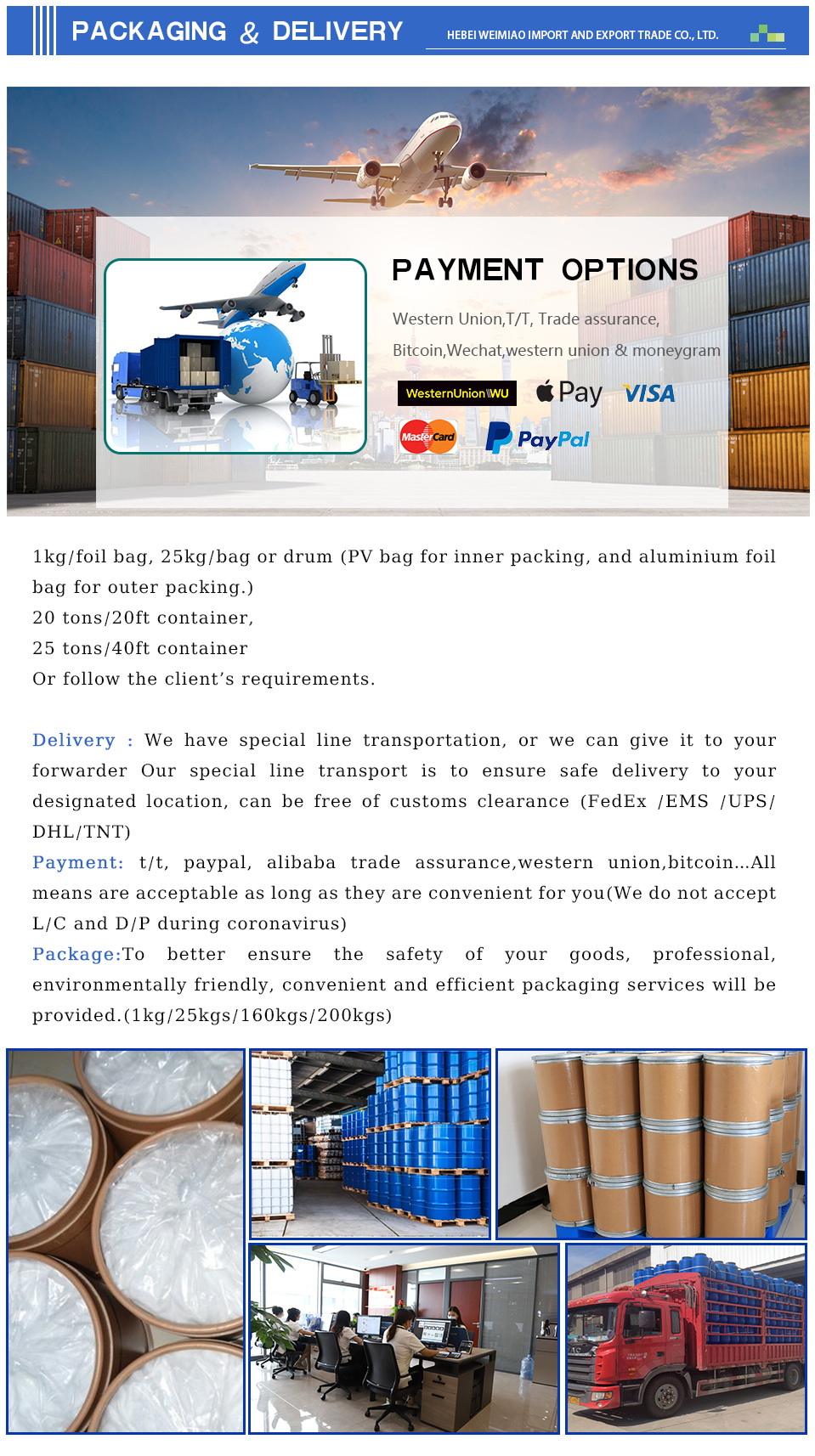
API failure modes often stem from degradation, contamination, or physical changes. Degradation pathways include hydrolysis, oxidation, photolysis, and isomerization. Hydrolysis is accelerated by moisture, while oxidation is promoted by oxygen and light. Impurities introduced during manufacturing or storage can compromise API quality. Physical changes, such as polymorphism transitions or agglomeration, can affect dissolution and bioavailability. Common failure analyses include HPLC impurity profiling, DSC thermal analysis, and XRPD phase identification. Preventative maintenance includes regular equipment calibration, cleaning validation, and environmental monitoring. Storage conditions (temperature, humidity, light exposure) must be controlled to minimize degradation. A robust change control system is essential to assess the impact of process modifications on API quality. Failure investigations should employ root cause analysis (RCA) methodologies to identify the underlying factors contributing to the failure. Corrective and preventative actions (CAPA) must be implemented to prevent recurrence. Stability studies are critical for determining the API’s shelf life and establishing appropriate storage conditions. Proper packaging materials (e.g., moisture-proof containers) are essential to protect the API from environmental factors.
Industry FAQ
Q: What is the significance of polymorphism in API manufacturing, and how is it controlled?
A: Polymorphism significantly impacts solubility, dissolution rate, and bioavailability. Different polymorphs of an API can exhibit vastly different performance characteristics. Control is achieved through careful selection of crystallization conditions (solvent, temperature, cooling rate), seeding techniques, and monitoring of the crystalline form using XRPD. Regulatory guidelines require thorough characterization of all polymorphs and justification for the chosen form in the final drug product.
Q: How are impurities controlled during API synthesis and purification?
A: Impurities are controlled through stringent process control, validated analytical methods, and purification techniques such as crystallization and chromatography. Impurity profiles are established, and limits are set based on safety considerations and regulatory requirements. Residual solvent levels are also carefully monitored and controlled using GC.
Q: What are the critical process parameters (CPPs) that require tight control during API manufacturing?
A: CPPs vary depending on the specific API and manufacturing process. Common examples include temperature, pH, reaction time, mixing speed, and reagent ratios. These parameters are identified through process risk assessment and are monitored and controlled within defined ranges to ensure consistent product quality.
Q: What role does the ICH Q7 guideline play in API manufacturing?
A: ICH Q7 (“Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients”) provides comprehensive guidance on GMP requirements for API manufacturing. It covers all aspects of production, from raw material sourcing to packaging and labeling, and is widely adopted by regulatory agencies worldwide.
Q: How do you validate the cleaning process of API manufacturing equipment?
A: Cleaning validation involves demonstrating that the cleaning process effectively removes residual API and cleaning agents to acceptable levels. It is typically performed using worst-case scenarios and utilizes analytical methods with sufficient sensitivity to detect trace contaminants. Validation protocols, reports, and re-validation schedules are crucial components of the process.
Conclusion
The manufacturing of pharmaceutical APIs demands meticulous attention to detail, robust process control, and unwavering adherence to regulatory standards. Understanding the underlying material science principles, potential failure modes, and critical performance parameters is essential for ensuring the quality, safety, and efficacy of drug products. Advancements in continuous manufacturing technologies and analytical techniques are driving improvements in API production efficiency and quality control.
Future trends will likely focus on sustainable manufacturing practices, improved supply chain resilience, and the development of novel APIs to address unmet medical needs. Continued investment in research and development, coupled with a commitment to continuous improvement, will be critical for maintaining the integrity of the pharmaceutical supply chain and protecting public health.